
Bad Drugs Get Pulled Fast
Safety issues usually get identified and acted on quickly

Many people are “unsure” or “hesitant” about different medications because they’re afraid of long-term harms, side effects that might only crop up after years of use. People commonly believe that the majority of those who take certain drugs will eventually pay some price. This view is based on a misunderstanding of risk—a misunderstanding that epidemiologists have an answer to.
The epidemiologists’ answer is person-time. Person-time is the total amount of time at risk that all individuals contribute to the data when they’re in a defined state of exposure, like taking an antibiotic or living with a surgery, summed across people. Person-time is the denominator used to compute incidence rates that comparable groups can expect to face over different follow-up lengths with some exposure.
To make how this works even clearer, let’s consider an example with device-time instead of person-time.
A data center installs 10,000 hot-swappable server fans and tracks them for reliability. Follow-up (i.e., device-time) starts when the fans are installed and accrues when they’re powered and in service. Device-time measurement stops at the first of: failure, elective replacement for an upgrade, server decommissioning, loss of telemetry, or the end of study. All of this is treated as a source of censoring if the thing that cuts off our observation is not some sort of device failure.
Over eighteen months, the fleet of fans contributes 96,200,000 fan-hours. There are, say, 700 failures (tachometer reading below threshold, seized rotor, etc.) in that time. That means that the failure rate is 700/96,200,000 = 7.28e-6 failures per hour. 7.28 failures per million fan-hours translates down to 7.28e-6 * 8,760 (hours in a year) = 0.065 failed devices per year, or 64 failed devices per 1,000 device-years. If the risk of a device failure is roughly constant over time1, then the mean time to failure is 1/7.28e-6 = or more than 137,000 hours (>15 years!). That’s the expected average time a single new fan should run for before it fails once. The median time to failure is ln(2)/7.28e-6, or about 95,000 hours, which means that half the units will fail by then.
Once we have the mean time to failure in hand, we can derive other facts about our fan fleet. For example, we can extrapolate that the annual failure rate suggests about 6% of units fail each year on average. By the mean time to failure, the expected fraction failed gives us an expectation that, after our ~137,000 hours, the first ~6,300 fans should have had a failure.
These extrapolations are going out more than a decade, but we only collected data for one-and-a-half years. How’s this possible? Because there is a distribution of time to failure. Some number of units will reach that failure point earlier, some later. But by studying large numbers of fans, we can figure out parameters for that distribution. This is how manufacturers find out about things like the reliability of lightbulbs, hard drives, LCD screens, and more. They don’t have to wait ten years to figure out that a television is likely to last you ten years—they can prove it definitively by letting time pass, but they don’t really need to—because they have access to a lot of television sets and that gives them a lot of device-time to extrapolate with.
The same thing holds with people. If you expose billions of people to, say, a vaccine, then you can start gathering billions of person-years of data right away. If there’s a major risk from the vaccine, then you’ll probably know it very quickly because people vary in how they respond to it and you have so much data to work with.
Epidemiologists lean on person-time for exactly this reason: it translates “how many people used a drug, and for how long?” into statistical power. With thousands, millions, or billions of people exposed to some drug, any severe harm that truly affected a large share of users would create a huge absolute event count. Even a 1% annual risk translates to tens of thousands of extra hospitalizations per year at the exposure scale of, say, GLP-1RAs, or SGLT2is, or acetaminophen, or immunotherapy or much else—far beyond normal background variability. A simple yardstick is the Poisson “rule of three”: if an outcome is never (or scarcely) seen over N patient-years, the upper-bound of the 95% confidence interval on its annual risk is roughly 3/N—vanishingly small when N is in the millions. That logic doesn’t prove there’s no risk to something per se, but it makes mass, severe harms extremely unlikely.
Crucially, we don’t rely on one data stream to figure out if something is risky. Multiple surveillance systems—pre-approval trials, spontaneous reports, active surveillance (e.g., in claims data, across EHR networks), and independent case-control and cohort studies—are continuously used to scan for spikes against expected background rates—safety signals. If a large, population-wide hazard existed, you’d see consistent, time-linked elevations across geographies, datasets, and comparators; you’d see dose and time-since-start gradients; and signals would persist after bias controlling techniques, like using active comparators, new-user designs, negative controls, and so on. In this context, the absence of large, reproducible risk patterns after a large amount of exposure is itself informative, it is evidence for the null.
What we see with most approved drugs today is the typical mature safety profile: uncommon, mainly organ-specific risks, often concentrated in subgroups or early in treatment, with small absolute risks that labeling and clinical monitoring can manage. That pattern is incompatible with a harm that severely affects most users a long time down the line. In fact, there has never been any drug like that. Reasonable caveats remain—very rare events, long-latency outcomes, or interaction-specific risks warrant continued follow-up of different drugs. But those categories, by definition, don’t produce the kinds of high-prevalence, severe toxicity that would have already shown up if it was a problem for many drugs, given today’s scale of collected person-time.
Now, let’s get into some empirical details.
The standards to get drugs withdrawn are often very low. The majority of drug withdrawals globally are based on anecdotal evidence—in the form of case reports—rather than on the basis of any sort of systematically gathered or causally informative evidence.2 This means that even weak safety signals are often sufficient to get a drug taken off the market. Despite that,

Globally, the time between the first reported adverse reactions and eventual withdrawals was about six years between 1950 and 2014. In the developed world and closer to the modern day, withdrawals tend to happen even more rapidly. For example, Health Canada’s median time between approval of a drug and its eventual withdrawal was 3.48 years for drugs approved between 1990 and 2009, as of 2014.3 For all FDA approvals between 1975 and 1999, half of the eventual withdrawals by 2000 happened within two years of approval. Even the FDA’s accelerated approval program for oncology drugs—intentionally lax approvals—had a median withdrawal time of just 4.1 years between the program’s inception in late 1999 and mid-2023. Similarly, the European Medicines Agency’s equivalent—Exceptional Circumstances and Conditional Approval—performs no worse in terms of withdrawals than the typical approval pathway.
Pharmacovigilance—meaning the science and activities pertaining to detecting, assessing, understanding, and preventing adverse effects and ensuring efficacy of medicines—has improved over time. In the FDA’s accelerated approval program for oncology drugs, for example, the median time for drugs to be converted to a regular approval declined from 4.3 to 2.3 years when contrasting the 1992-2013 and 2014-24 periods. The efforts to confirm accelerated approvals also increased, with the number undergoing confirmatory studies at the time of accelerated approval increasing from 63% to 85%. More importantly, the withdrawal delay declined from 9.5 to 3.2 years. Looking at the program as a whole, withdrawal delays have clearly declined:
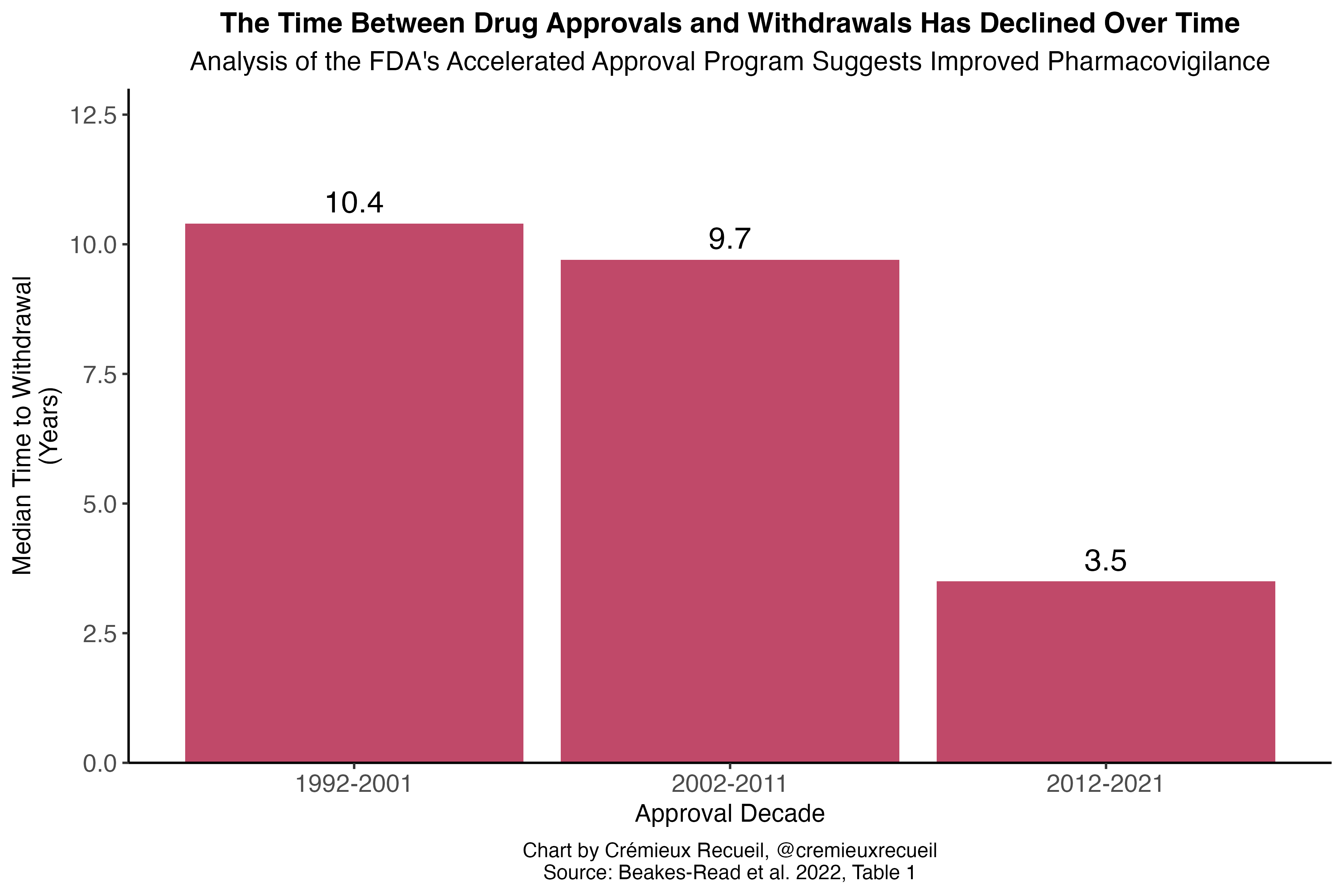
Pharmacovigilance is something that regulators take seriously. They do monitor for drug risks, support studies to evaluate said risks, improve risk detection methods and promote risk detection tools, and they frequently suggest withdrawals arguably too aggressively. Consider the case of the decongestant phenylpropanolamine (PPA).
PPA was pulled from the market because of a very small risk of stroke among women who abused the drug for weight loss. The statistical evidence for PPA harms is poor because the supposed acute effect—stroke—is so rare, at just 1 in 107,000 women who used the drugs. The studies justifying the risk claim were also of the case-control variety; participants were asked to volunteer any PPA usage they could remember, but memory is impacted by having a stroke, making the results unreliable. Nevertheless, the signal seems plausible. It weakly replicated, and seemed to do so solely among women using the drugs at a high dosage for the purpose of weight loss.
But the risk might not have even been due to PPA itself. PPA interacts with caffeine in a pharmacokinetically unexpected way: in animals and in people, when PPA is taken with caffeine, plasma caffeine levels skyrocket because it inhibits the enzyme CYP1A2; blood pressure follows suit. Women looking to lose weight frequently consumed both PPA and caffeine in excessive quantities, and the skyrocketing plasma concentrations as a result of the interaction made for frequent toxicity concerns. Some formulations of PPA for weight loss (Westrim, Acutrim, Dexatrim, etc.) even included caffeine, making the risk that much worse.
Because there were alternative decongestants available, the FDA and other agencies had little reason to keep PPA on the market given the possibility of risk. But, oddly, there’s no reason to believe that PPA without caffeine is even risky. Therefore, there’s a case to be made that the FDA made the wrong choice, that it acted too aggressively, and pulled PPA from the market when it shouldn’t have been removed at all. But, because caffeine is so common, there’s also a decent case to be made for removing the drug from the market, since it can be hard to avoid risk. Who’s to say?
Bad drugs tend to get pulled quickly, and today that’s more true than ever. When drugs genuinely have side effects that outweigh their benefits, it’s easy to tell. As a result, there are practically no examples of drugs or vaccines that are like that which have been kept on the market for any appreciable amount of time. But somehow people believe otherwise. They have an undeservedly negative view of pharmaceuticals—for which the total withdrawal rate is only about 1-5%, and not even close to all of which is due to safety issues—and of regulators—who are actually generally competent.
Why? Who’s to say. I can speculate that it has to do with unscientific thinking, conspiracist ideation, or plain-old confusion, but at the end of the day it doesn’t matter. It is what it is, so all we can say is that, if people want to talk a big game about risk, they need to get serious, because our informed prior should be that regulators already are.
This is not a critical assumption, nor is it always used in practice. I am just using it for the sake of explaining this simple model.
Though I focus on withdrawals here, black box warnings, REMS, other restrictions, etc. are the means used to handle most safety issues. Regulators have gotten quicker and more expansive about issuing warnings, but they also often put out warnings too hastily. For example, consider the warnings about height on ADHD drugs.
For each cited article like this, updating the endpoint would have a meager effect.
> If the risk of a device failure is roughly constant over time,
This is the critical assumption that sometimes does not hold with drugs. Lets take for example tobacco. If in group A we have 1000 people who smoke 1 pack a day for 1 year, and in group B we have 50 people who smoke 1 pack a day for 20 years, well, these two groups have the same 1000 person-years of exposure. But we aren't going to see the same number of adverse health outcomes in the two groups. We are going to see more adverse health outcomes in group B, because the harmful effects of tobacco build up over time. I think we should at least take seriously the risk that something similar happens with any new drug.
You make a good point that fear of new medications is generally overstated - Thalidomide and DES not withstanding. But there certainly are drugs with cumulative ill effects that come from long term use - lithium which harms the kidneys and antipsychotics (especially first gen) that produce Tardive Dyskinesia, come to mind. Probably the most important issue is drug regulation is the approval of drugs (against placebo) that have little benefit over the placebo and certainly no benefit versus other cheaper drugs that are already available. Add to these sham "long acting" reformulations to extend patent rights and you have a drug industry that does not do itself proud most days.