
Statins and Strength
Does lowering your LDL and saving your heart mean sacrificing muscular strength? No.

This was a timed post. The way these work is that if it takes me more than one hour to complete the post, an applet that I made deletes everything I’ve written so far and I abandon the post. You can find my previous timed post here. Everything beyond the paywall was not limited by the timer.
In my previous article, I used statins to elucidate the nocebo effect, where patients believe they’re being treated and that treatment causes side effects, so they begin reporting symptoms, even with placebo drugs. Essentially, fear is responsible for many of the reported medical side effects. This is also true for other drugs and compounds like antidepressants, MSG, and even completely unreal ‘chemicals’.

In response to the specific revelation that most side effects of statins are due to the nocebo effect, several people recounted their anecdotes about statin side effects as a supposed rejoinder to what I said. Ironically, they were likely just demonstrating that they suffer from the nocebo effect. But I have to digress.
The most commonly reported anecdote was that statins made people weak, that they materially impacted their ability to lift weights effectively. I’m sure some people believe this has happened to them, but it’s implausible, as there’s no real mechanism for this to happen. The most likely way it would happen—if it does, in fact, happen—would be through some sort of psychological mechanism. Luckily, we don’t have to go back-and-forth with combatting anecdotes or N-of-1 self-experiments of unknown reliability; instead, we can just review the literature on this subject!
The Literature
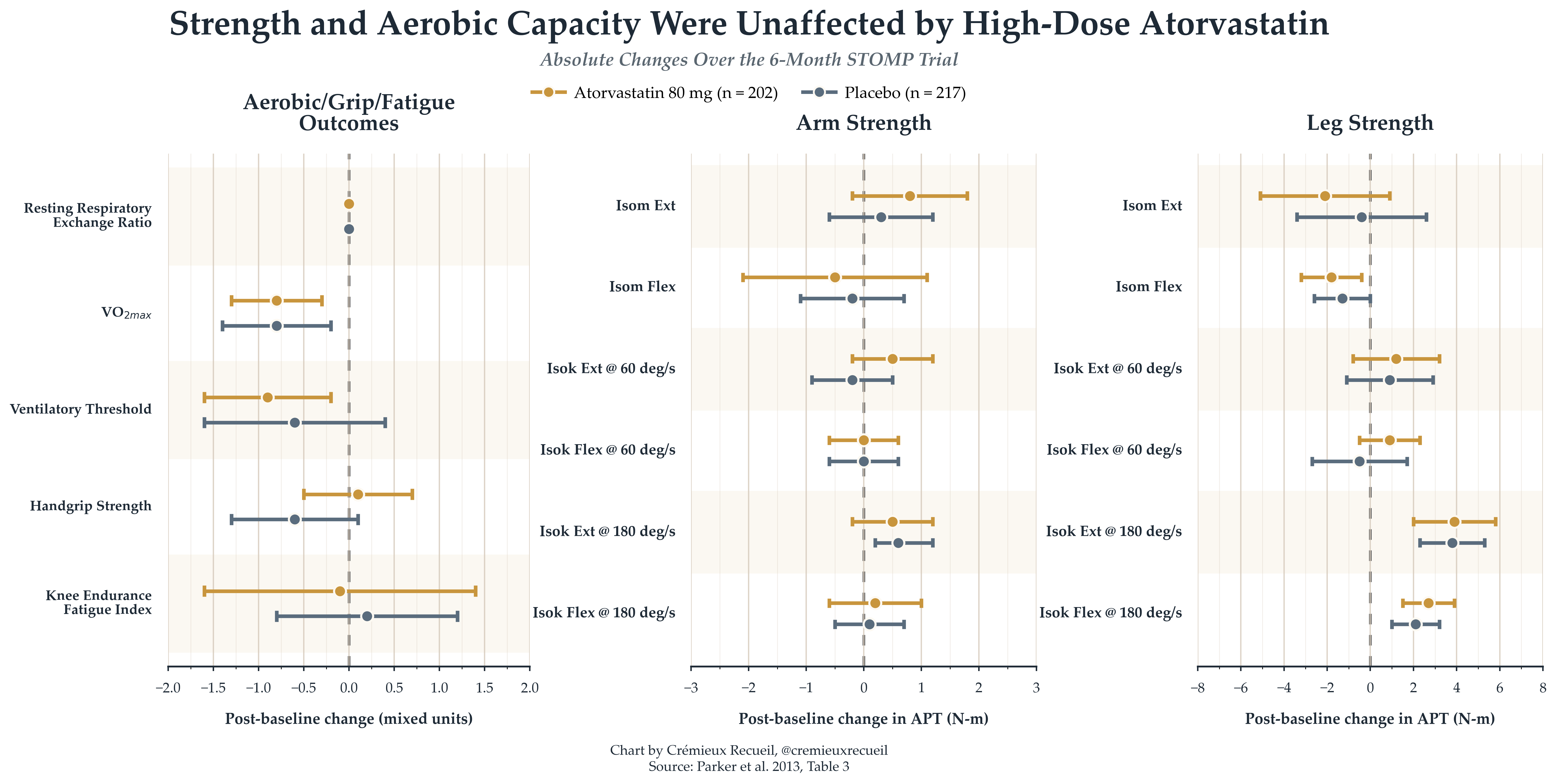
STOMP: The biggest and best trial on statins and strength is The Effect of STatins On Skeletal Muscle Function and Performance, or STOMP, a 2013 trial in which 420 participants who had never taken statins were randomized to either a placebo or high-dose (80mg) daily atorvastatin for six months. Participants were assessed on strength and aerobic capacity at the end of the beginning and end of the intervention, and the changes that happened during the trial were practically identical:
To make the placebo-intervention difference clearer, we can look at contrasts:
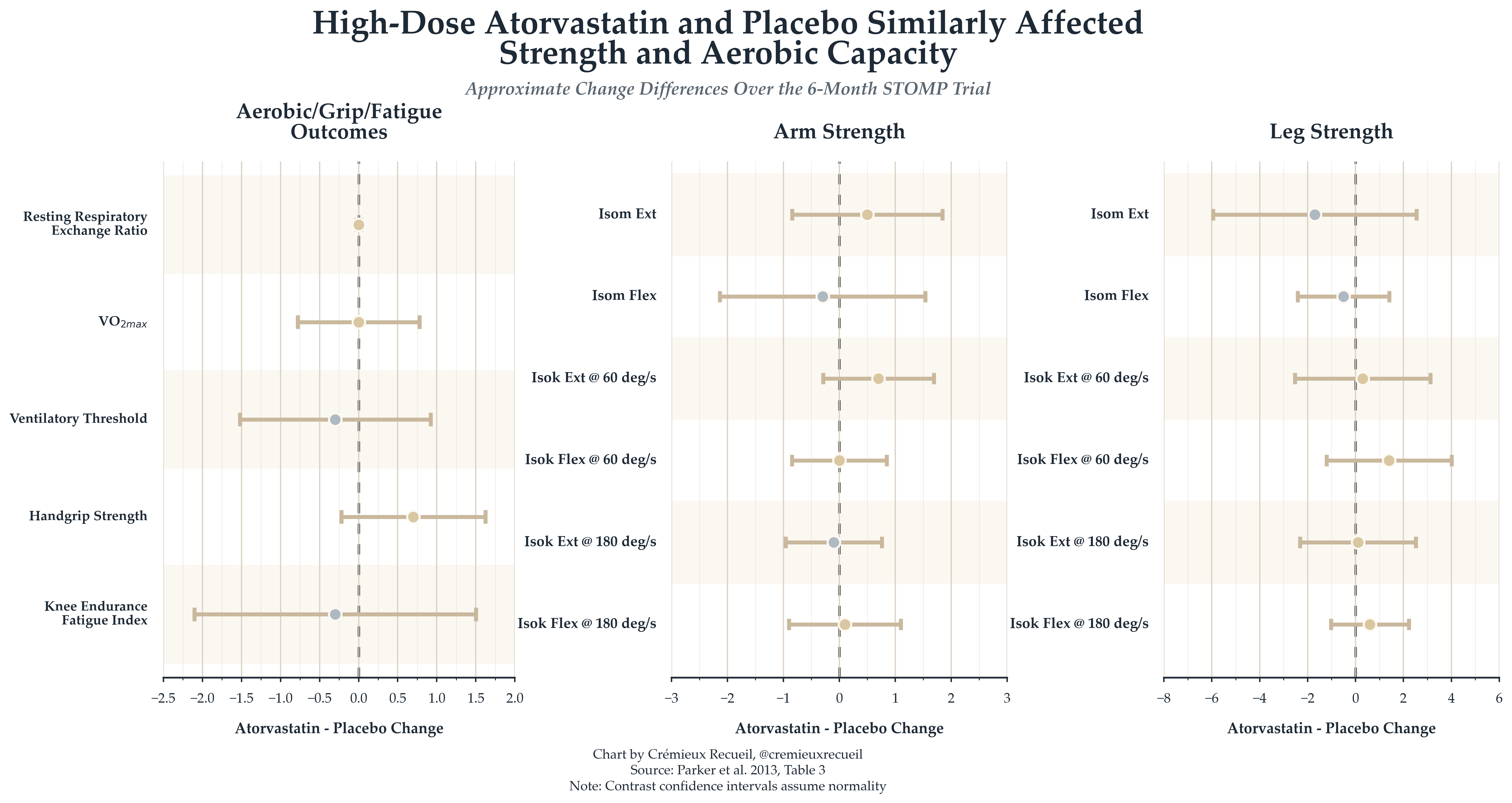
Essentially nothing happened, and this was the case even though the authors seemed to be biased in favor of statins being harmful. We know this because of two reported results.
The first is for creatine kinase—a useful, but blunt marker of muscular damage. Creatine kinase has poor specificity for magnitude and meaning; reviews have stressed that it isn’t a good predictor of maximum voluntary contraction loss or other aspects of functional damage and older papers have noted that peak creatine kinase doesn’t seem to even correlate with soreness. This is supposed to be a measure of muscle damage, but it’s more a measure of what it actually is, which is muscle cell content leakage into the blood, which is only alarming at highly elevated levels. In any case, creatine kinase does not seem to progressively increase on statins, and in this trial and others, it only increases slightly and not generally clinically significantly. In fact, in STOMP, creatine kinase levels didn’t predict lower muscular performance.
The second is for myalgia—muscle pain—, which I covered in my previous article. The authors reported that myalgia was more common in the statin group than in the placebo group (n’s = 19 and 10, respectively). But, curiously, myalgia subjects had worse muscular performance on 5 of 14 measures and placebo participants with myalgia had lower performance in 4 of 14 variables—no significant difference.
The bias evidence comes from how these results were reported. The creatine kinase result was only reported with numbers on creatine kinase for the statin group, so we can’t calculate the interaction effect. This result is also overhyped. The myalgia result is more severely misreported, as it’s just entirely incorrect. While there were more myalgia reports in the statin group, this difference was not significant: not how the authors reported it, and not actually. The authors reported that the difference was significant, with a p-value of 0.05. But, significance has to do with the p-value being below a given threshold, not at it. But their reported p-value wasn’t even correct! A Pearson χ2 test without continuity correction returns p = 0.05495, which the authors could round down to 0.05 for their incorrect report. But this isn’t even robust! With continuity correction—which isn’t needed—the p-value becomes 0.084, and a two-sided Fisher’s exact test returns 0.082. A one-sided test would be marginally significant, but it is not justified, as the authors wrote that they had no good prior:
Many clinicians believe that statins cause muscle pain, but this has not been observed in clinical trials, and the effect of statins on muscle performance has not been carefully studied.
In an earlier (2008) study of older adults by Traustadóttir, Stock and Harman, the same result was achieved with high-dose simvastatin over a period of twelve weeks. The authors also found suggestive evidence that intramuscular CoQ10 wasn’t meaningful for these outcomes. But, this study was small (n = 10) and had no control group. Annoyingly, most other studies don’t deal with real functional outcomes, but instead, with biomarkers of dubious validity and which are less likely to show changes in studies that aren’t directly talking about them—a pattern indicating publication bias.
The main problem with STOMP is that it’s not revelatory about the response to exercise, so much as the stationary (i.e., not actively trying) effect on aerobic and muscular capacity. While those are not affected, might people who are on statins progress more slowly?
Coen et al. (2009) found that training benefits can still occur for people undergoing rosuvastatin treatment, but this had no non-statin-using control group.
Mikus et al. (2013) actually had a control group for a study of exercise versus exercise plus simvastatin, and they seemed to find that simvastatin blunted the increase in cardiorespiratory fitness indicated by VO₂ₚₑₐₖ, such that this increased 1.5% instead of 10%. But, this study can’t be trusted. For one, there was no placebo, so the study is vulnerable to placebo/nocebo effects. For two, several reported p-values are impossible or improbable. For example, the interaction by group is reported differently across Figure 1A and 1B, and the p-value reported for the group difference—p < 0.005—is impossible. The actual p-value is marginally significant, at p = 0.01-0.02. For three, several outcomes require implausibly large pre/post correlations. For example, to hit reported p-values for weight change within the exercise group or between groups, to hit them for fat mass and lean body mass changes within the exercise or exercise plus statin groups, and for the latter outcome, between groups, as well as for body fat percentage within the exerciser group, the correlations would have to be 0.991, 0.985, 0.944, 0.993, 0.987, and 0.968. This is all highly implausible, and it’s matched by the need for a >0.95 VO₂ₚₑₐₖ correlation over time if Figure 1A is to be believed. None of this is particularly believable.
Slade et al. (2021) had two studies, the latter of which was not so much an exercise RCT as a post hoc analysis of statin users in a twelve-week exercise RCT. The former study involved assessing the effects of four weeks of high-dose atorvastatin on practically irrelevant outcomes like the rate constant of phosphocreatine recovery (kPCr) and muscle pH, which are not functional. In the latter study, the VO₂ₚₑₐₖ response to exercise was -1.9 points for non-statin users in the control group vs. -0.8 for control group statin users and +1.7 for non-statin users in the exercise group vs. +2.7 for exercise group statin users. With corresponding exercise effects of 3.6 vs. 3.5, the difference is essentially zero and statins do not appear as a moderator. In fact, with a correlation over time of r = 0, the interaction p-value would be 0.98, and with r = 0.9, it would be 0.96. In the same year, Allard et al. (2021) found no differences in strength improvements between supposedly symptomatic, asymptomatic, and non-statin-using subjects who completed a twelve-week physical training course.
Reviews have consistently emphasized that there does not seem to be much in the way of evidence for statin-related physical performance harms, but the evidence base is small and it’s not as if minor harms can be excluded from possibility. In a review of the small number of RCTs out there, we got this result, speaking to how imprecise the literature is:
Of the 3 RCTs providing qualitative SAMS results, 19 (9%) out of 220 participants reported SAMS on exercise+statin and 10 (4%) out of 234 reported SAMS on exercise+placebo. There was no difference between exercise+statin vs exercise+placebo for maximal oxygen consumption (d=−0.18; 95% CI, −0.37 to 0.00; P=.06) or creatine kinase after short-term exercise (d=0.59; 95% CI, −0.06 to 1.25; P=.08).
A Simple, Long-Term Test
A much easier way to check if statins or other LDL-lowering medications cause long-term muscular or aerobic harms is to use Mendelian Randomization. The gene corresponding to statins as a drug category is HMGCR, and the effects of variants in it mirror the effects of the drug exceptionally closely, making them excellent means of projecting the lifelong effects of taking a statin. For my purposes, I’ll be scaling them like the Cholesterol Treatment Trialists’ (CTT) Collaboration does the effects in statin RCTs: in terms of the effect of a 1 mmol/L lowering of LDL cholesterol.

To index the effects of statins rather than LDL-lowering medications more generally, we’ll be using SNPs in HMGCR. I’ll use its canonical instrument, and for robustness, a strict cis instrument, a loose set of spaced cis instruments, and a relaxed set of plausible cis instruments.1
For lifespan outcomes, I used the Zenin et al. healthspan GWAS, the Park et al. lifespan GWAS, and the Timmers et al. parental lifespan GWAS. For aerobic outcomes, I used the Hanscombe et al. cardiorespiratory fitness slopes and VO₂ₘₐₓ, the Klimentidis et al. physical acceleration measures, for maximum workload and heart rate as well as heart rate achieved during the U.K. Biobank’s fitness test, I used the Neale UKB PheWeb.
For strength and muscle outcomes, I checked hand grip strength for left and right hands using the UKB Neale 46 and 47 entries, GCST90014019, the Pan-UKB meta and meta HQ, GCST90007526, GCST90007529, Abbondanza et al.’s childhood grip strength GWAS, I checked appendicular lean mass with GCST90000025, lower limb strength in older Japanese adults with GCST90319502, the frailty index with GCST90020053, stair climbing frequency came from UKB Neale 943 and the Pan-UKB sources, while usual walking pace was UKB Neale 924 and also those Pan-UKB sources. I was able to make the left/right hand grip strength, lower limb strength, and appendicular lean mass results age-annotated as well, going from 40-69 for hand grip strength and appendicular lean mass and 60 and onwards for low grip strength in EWGSOP/FNIH and lower limb strength.
I also checked bone mineral density and did so across different ages with GCST005345 (kids), GCST005344, GCST005346, GCST005350, GCST005349 (elderly adults), and GCST005348 (all ages). And finally, I decided to additionally check effects on diabetes risk (Neale 2443; also HbA1c as Pan-UKB 30750), body fat percentage (23099), BMI (21001), and standing height (50), purely because they’ve been discussed with reference to statins. I also added results for bempedoic acid (ACLY), ANGPLT3 inhibitors, APOB inhibitors, CETP inhibition, the LDLR pathway, MTTP inhibition, ezetimibe (NPC1L1), and PCSK9 inhibitors below the paywall, plus GLP-1-stimulated insulin secretion, dementia, memory loss, and ICD10 R41 cognitive symptoms as outcomes.
And now, let’s get onto the outcomes in the order we brought them up.
For healthspan, the effect for the increasingly expansive sets of HMGCR instruments is +0.37, +0.07, +0.59, and, finally, +0.93 years with only the last result being statistically significant (0.26-1.59). For lifespan, the results are +2.30 (ns), +5.05 (1.25-8.85), +7.60 (5.41-9.80), and +7.36 (6.09-8.64) for men and +3.18 (ns), +4.23 (ns), +4.69 (2.04-7.34), and +4.23 (2.70-5.77) for women. Most interestingly, the results for parental lifespan were +0.89 (ns), +0.98 (ns), +1.45 (0.48-2.42), and +1.44 (0.89-1.99).

Cardiorespiratory slopes increased by 0.035, 0.039, 0.031, and 0.029 points, but only the last result was significant (0.012-0.046). VO₂ₘₐₓ was somewhat higher by 0.057, 0.065, 0.058, and 0.090, but only the last result was significant (0.011-0.170). The results for wrist accelerometer activity (0.182, 0.506, 0.502, 0.370) and average acceleration (0.264, 0.807, 0.582, 0.500) were universally positive in direction but all nonsignificant. Maximum workloads were higher by a nonsignificant 0.041 for the canonical specification and lower in all the rest (-0.094, -0.140, -0.136), with the latter two results being significant (-0.277 to -0.003; -0.061 to -0.212). The maximum heart rate achieved was somewhat lower (-0.168, -0.259, -0.295, -0.270) with the latter three results being significant (-0.508 to -0.010; -0.450 to -0.140; -0.354 to -0.185). Rates of achieving target heart rates were not significantly lower (-0.009, -0.040, -0.021, -0.010).
Results for left (0.003, -0.002, -0.006, -0.012) and right (0.041, 0.020, 0.012, 0.005) grip strength were consistent with one another, but not significant. Results for childhood dominant (0.392, 0.404, 0.376, 0.540) and non-dominant (0.338, 0.346, 0.279, 0.470) grip strength were also consistent with one another, and the last results in each set were significant (0.233-0.848; 0.162-0.779). The results for lower grip strength were aligned but nonsignificant. Appendicular lean mass was consistently higher with lower LDL (0.078, 0.094, 0.114, 0.100) and all of these results were significant (0.008-0.148; 0.018-0.169; 0.070-0.157; 0.074-0.126). Knee extension strength was also higher (0.043, 0.259, 0.409, 0.292), but not significantly so.
The frailty index was lower (-0.152, -0.157, -0.139, -0.145), and all of these results were significant (-0.274 to -0.029; -0.288 to -0.026; -0.221 to -0.057; -0.190 to -0.100). Stair climbing frequency was lower (-0.089, -0.111, -0.106, -0.102), and the latter two results were significant (-0.185 to -0.028; -0.145 to -0.058). Usual walking pace was consistently lower (-0.092, -0.107, -0.113, -0.112) and all of these results were significant (-0.148 to -0.037; -0.167 to -0.047; -0.149 to -0.076; -0.132 to -0.091).
Bone mineral density was consistently not affected, as you’d expect. Diabetes had nonsignificant odds ratios of 0.998, 1.004, 1.001, and 0.999. HbA1c was elevated by 0.167%, 0.162%, and 0.155% (twice), and all such elevations were significant (0.110-0.225; 0.101-0.223; 0.120-0.190; 0.135-0.175). Body fat was a bit higher with lower LDL (0.90pp, 1.05, 1.17, 1.16), and this was significant (0.61-1.18; 0.74-1.35; 1.00-1.34; 1.06-1.26). BMI was also somewhat higher (0.83, 0.95, 1.02 twice), and this was significant (0.61-1.04; 0.72-1.18; 0.90-1.15; 0.95-1.10).
Finally, as a sort of negative control given the total lack of any plausible mechanism, standing height was somewhat higher for people with lower LDL (0.11cm, 0.30, 0.68, 0.56), and these results were significant for the latter two outcomes (0.34-1.03; 0.37-0.76). Also, among the more obvious outcomes were a 27% reduction in ischemic heart disease, 19% fewer myocardial infarctions, 37% less coronary atherosclerosis, and 36% less peripheral artery disease, plus 62% lower risk of vascular dementia—basically tautological outcomes, making them somewhat meaningless to check beyond their utility in verifying the method produces the right results where we expect them.
In short, the lifelong effect (into people’s early elderly years) of 1 mmol/L lower LDL due to statins is generally beneficial, with no signs of reduced strength or muscularity, somewhat larger glycemic effects than are observed in trials—as expected given their much shorter duration—, and near-zero, clinically meaningless effects for most things, alongside considerable life extension and greatly reduced cardiovascular risk.
